Update, 1 March 2016: pubmed.mineR has recently been updated a couple times. Since the most recent update (1.0.5), the old API works again, so the code on this page will work. However, I have not been able to reproduce the (wonderful) results that I had before the recent updates to pubmed.mineR. Use with caution.
Something a bit different today: a little manual for using a package for the R programming language for text mining.
Pubmed.mineR is a “library” for doing text mining from the PubMed/MEDLINE collection of documents. PubMed/MEDLINE contains references to about 23 million articles in the domain of biomedical science, broadly construed. It was released with documentation for the various and sundry methods that it provides, but no manual. This blog post is an attempt to put together a basic manual for using it, with some code examples. Pubmed.mineR was written by Jyoti Rani, Ab Rauf Shah, and Srinivasan Ramachandran. You can find an article about it here, and some documentation here.
First, you need to have the input data in the right format. Here’s a screenshot from one of the authors, Ramachandran, showing how to do a manual query and then save the results in the proper format:

Here is some sample R code for the library:
# import the library
library(pubmed.mineR)
# read in the abstracts
abstracts <- readabs(“pubmed_result.txt”)
abstracts is an object of type S4. This is a kind of class used for doing object-oriented programming in R. abstract is printable with print(abstracts). An S4 object stores its data in slots. To understand slots in R, try this web page: http://stackoverflow.com/questions/4713968/r-what-are-slots.
The abstracts class has the following slots:
- Journal: This returns a vector of the names of the journals for each publication in the whole collection.
- Abstract: This returns a vector of the abstracts for the whole collection.
- PMID: This returns a vector of the PMIDs for the whole collection.
(It’s worth noting that the elements of some of these vectors have some oddities. For example, when you get the vector of titles, you’ll notice that each one is prefaced with the number of the element of the vector. I suggest looking at these outputs closely, as I’m sure that I haven’t picked up on all of these oddities.)
So, this line of code will get you a vector of the PMIDs (some columns trimmed from the output for readability):
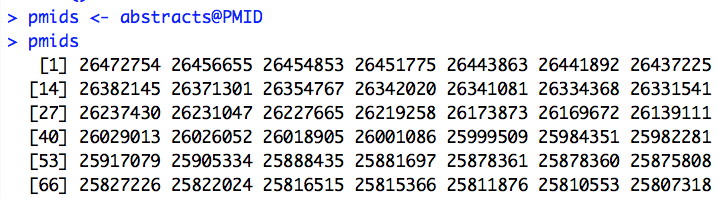
Once we’ve got a PMID for an abstract, one thing that we can do with it is send it to PubTator. Once we can do that, we can get access to lists of the genes, mutations, diseases, and chemicals that are mentioned in the abstract. (Some columns of output omitted for readability.)

These lines of code will get you access to the rest of the stuff in the PubTator results:
pubtator_output$Genes
pubtator_output$Mutations
pubtator_output$Diseases
pubtator_output$Chemicals
pubtator_output$Species
It’s pretty common to want to iterate over all of the sentences in an abstract. You can do that by getting a vector of the sentences with the SentenceToken() method. It has to be passed a character string, so you’ll want to pass it an element of the vector of abstract bodies that you get from abstracts@Abstract:

A question that immediately arises is whether you can pass individual sentences to the PubTator function. I haven’t had good luck with that–it always seems to return “No data.” So, I guess that I would try running pubtator_function() on the whole abstract, and then search individual sentences for the things that pubtator_function() returns with a regular expression or substring function or something.
This doesn’t exhaust everything that you can do with pubmed.mineR, but it should be enough to get you started. Good luck, and if you figure out how to do something cool with it that I haven’t talked about, please tell us in the comments!

where is pubmed_result.txt ????
LikeLiked by 1 person
You have to create it. It contains the results of your PubMed/MEDLINE query. See the screen shot at the top of the post for information on how to create the file.
LikeLike
I have downloaded the pubmed_result.txt file into R using the File/open file option but when I use searchabs, to search for objects in file, it does not recognize the file. Any recommendations?
LikeLiked by 1 person
I suggest contacting the authors of the package. If you get an answer that works, it would be super-helpful if you could post it here!
LikeLike
Go to the directory where you have downloaded the “pubmed_result.txt” file, then use the following command to bring in the file in the R environment: abstracts <- readabs("pubmed_result.txt")
LikeLiked by 1 person
Hello Colleen
You need to import Pubmed abstracts using readabs() function of pubmed.mineR.
LikeLike
It would be better to work with the xml file that Pubmed offers instead of the abstract text file
xmlreadabs() is the corresponding function to read the xml file
This should reduce inconsistencies mentioned ….
LikeLiked by 1 person
Yep, loading the abstracts from the xml file using xmlreadabs() worked much better for me. +1
LikeLike
Please see the tutorial here
https://docs.google.com/presentation/d/e/2PACX-1vRtv_5fmtW9x6W1_Sgvd2_3EkFZnTdxwnWgc-xxsVEnb_38B3DECLkMc_OKKvqwGQUHUt_qr2pJ3YRE/pub?start=false&loop=false&delayms=3000
LikeLike
can i extract abstract based on my pmids?
LikeLike
Yes, it’s the Abstract slot:
> Abstract: This returns a vector of the abstracts for the whole collection.
LikeLike